It is an amazing time for adeno-associated viral (AAV) gene therapies, as well as ex vivo and cell-based approaches to treat and cure thousands of diseases with unmet needs. For example, in November 2022, joining the previously launched Luxturna and Zolgensma, a third AAV vector gene therapy was granted FDA approval for the treatment of hemophilia B: Hemgenix (CSL-Behring, LLC). Hemophilia B is associated with congenital clotting Factor IX deficiency. Patients with this condition may receive periodic infusion of Factor IX to reduce the risk of severe and often life-threatening bleeding, but this continuous prophylaxis is expensive and requires patient compliance to the regimen to control the disease over the patient’s lifetime. Similar to its FDA-approved gene therapy predecessors, a single dose infusion of Hemgenix is often both corrective and curative.
At the time of press, there were 3 gene therapies and 26 cell therapies approved by the FDA, but there is a rich pipeline of hundreds of candidate therapeutics in all phases of clinical development and evaluation, across all platform modalities. Some notable approved therapies include the autologous cell-based Zynteglo (beta thalassemia, Bluebird Bio), topical gene therapy, Vyjuvek (dystrophic epidermolysis bullosa, Krystal Bio), Roctavian (severe hemophilia A (Biomarin), and Elevidys (Duchenne muscular dystrophy, Sarepta). In 2023, we also saw approval of Casgevy, an editing technology for beta-thalassemia and sickle cell disease (CRISPR Therapeutics). Among the hundreds of early stage clinical candidates, UX701, a gene therapy for Wilson’s disease (Ultragenyx) and RPL102 (Rocket Bio), a lenti-based ex vivo gene therapy indicated for Fanconi anemia are among the many candidates vying for late stage clinical and ultimately regulatory approval.
Creating a platform approach
With potentially thousands of genetic diseases treatable by gene therapies, the possibilities are nothing short of staggering and we can expect to see a need for higher manufacturing capacity and volumes in the future. For the present, however, these therapies typically target very niche patient populations and are not front-line treatments. Patients that are eligible for a gene therapy or that take part in a gene therapy clinical study are typically quite ill, face comorbidities, and may be experiencing side effects from standard of care treatments. For these reasons, speed to first-in-human examples for new therapies is crucial; delays in reaching the clinic can cost patient lives.
There is a perception that the development of a gene therapy is a complex process. In many ways this is true, but the development of processes and analytics, as well as the manufacture and release of product, can be simplified and standardized to a large extent by using a platform approach incorporating single use and scalable equipment. In a platform approach, time savings can be realized in all steps of the development and manufacturing processes; from project launch, to rapid and focused development studies, and engaging a templated transfer to the manufacturing and quality control (QC) groups. For example, a standardized process may include the use of consistent consumables, buffers, and media, which helps to simplify the supply chains, and means that materials can be stocked in advance. Developers can also benefit from templated master batch records and standardized test/release methods. Templated manufacturing records and platform release assays, for example, dramatically reduce the time to review and approve methods, as well as simplifying training, and reducing errors in GMP production. In short, the GMP, QC and quality assurance groups know what to expect, and are able to execute efficiently and with minimal error or delay, leading to faster and higher quality delivery of therapies to the patient.
An example of a platform approach is graphically described in Figure 1. The platform approach comprises, in part, early, well-documented production of pre-clinical viral vector lots in the non-GMP development laboratory. These materials are produced from a locked and (preferably) scaled process on equipment comparable to that used in GMP manufacturing. Vector lots for preclinical toxicology or efficacy may be produced in this way, as well as reference material for assay qualification to support GMP vector release for first-in-human studies. Raw materials should ideally be off-the-shelf and may include a well-performing clonal HEK293 production cell line, and off-the-shelf transfection reagent and plasmids. High quality plasmid is critical to a successful transfection, so extensive QC release testing is needed for these materials.
Essential to the platform approach is the inclusion of program-specific approaches for optimization. In this phase, process developers will need to conduct focused development (such as using small scale experiments in flasks/parallel bioreactors) on the variables most critical to productivity and quality/safety, such as transfection reagent selection and plasmid molar ratio. Here, in-process analytics are critical to verify expected vector titers and quality as early in development as possible; it is much easier to correct problems at the development stage than it is in manufacturing. Key in-process analytics include vector genome titer (ddPCR, dPCR), total capsid (ELISA), encapsidated hcDNA (qPCR), vector protein profile (SDS-PAGE), and fidelity of vector packaging/percentage of full AAV particles (analytical ultracentrifugation).
For a locked platform process, the corresponding process and release analytics should be locked in when manufacturing is reached. Representative release testing for both cell and gene therapies are summarized in Table 1. As is evident from the table, comprehensive safety and residual testing is needed to measure for vector quality, potency and safety. Encapsulated host cell DNA may present a significant regulatory and safety risk and must be measured and monitored closely. Adventitious viruses may be propagated during cell culture, so in vitro testing for these materials on crude harvest material is used to monitor upstream culture samples directly.
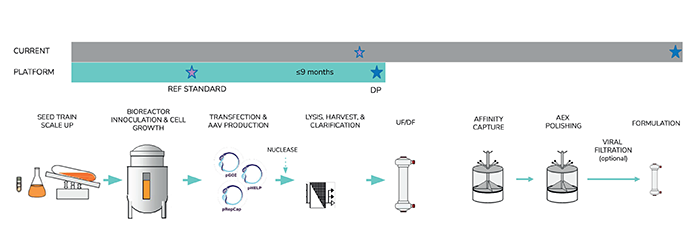
Figure 1.
The road to commercialization
During the development of any gene therapy, developers must keep their methodology focused on the ultimate goal of commercialization. During early development and initial clinical evaluation, it may be too early to make substantial investment in commercial activity pending early clinical outcomes. However, the foundation should be set in these early stages to enable seamless transition to late-stage studies and, in due time, regulatory submission. Platform methods would mean that the commercial teams are already informed of the key materials, which they can stock and qualify; batch procedures can be templated and staff trained; and release testing can be validated. AAV viral vector development and manufacturing will still vary between each project (even swapping a transgene in an otherwise-locked process may give differing yields and quality), but overall a platform approach will help the manufacturing and quality teams be more prepared. A robust change control process, and thorough training for operators as needed, can also be put in place in anticipation of minor adjustments to the platform.
Thousands of diseases with a genetic basis may be curable with gene therapy approaches. Current barriers include low vector yields and high cost of goods, but continued innovation will eventually bring viral vector development and manufacturing to the state of biologics: monoclonal antibodies are routinely produced in multi-gram quantities per liter using stable cell lines, perfusion and high-density culture. Catalent is already seeing growing use of platform approaches and expects these to be further optimized as industry experience with commercial gene therapies increases.
Overcoming the Immune Barrier
As well as the development of more platform-based approaches to development and manufacturing, other innovations are also being seen in the gene therapy space. Novel capsids, for example, can improve targeting to the correct tissue, as well as direct delivery (as opposed to systemic) to the target tissue or organ, reducing dosing requirements and potential toxicity.
Some of the greatest barriers to AAV-based theories are pre-existing immunities to the AAV capsid. The vector itself can also invoke both humoral and cellular deleterious responses that may cause inflammation or other toxicity. Further, in the current state, re-dosing with the same AAV vector is not possible due to the immunity generated at the first dose. The study of AAV-induced deleterious immune response and possible remedies is an area of tremendous, ongoing study. The literature is rich with reviews and studies of progress toward reducing or eliminating this immune-based barrier. Reduced dosing by improved targeting is one possible remedy, and this may be achieved by direct delivery of the vector to the correct tissue or targeting via modified capsid design (1). In another approach, scientists describe immune response derived directly from the AAV transcription and translation products. The authors contend that pathogen associated molecular patterns (PAMPS) may be modified to reduce deleterious immune response (2). Addressing immune-induced toxicity and inflammation further opens the door to this exciting area of new and curative medicines.
Newsletters
Receive the latest pharmaceutical news, personalities, education, and career development – weekly to your inbox.

References
- HCJ Ertl, “Immunogenicity and toxicity of AAV gene therapy,” Front Immunol., (2022). DOI: 10.3389/fimmu.2022.975803. eCollection 2022
- BA Hamilton, JF Wright, “Challenges Posed by Immune Responses to AAV Vectors: Addressing Root Causes,” Front Immunol., (2021). DOI: 10.3389/fimmu.2021.675897